The computerized representation of chemicals introduced in the previous chapter enabled computers to store chemical structures effectively. Scientists have used this ability to build databases of chemical information collected from hundreds of thousands of experiments. For example, experimentally determined physicochemical properties such as melting point, water solubility, pKa, and LogP can be assigned to each chemical, meaning there is no need to repeat the experiment to know that property. Another very informative type of data that can be assigned to chemical structures is their spectra produced from spectroscopic analysis. For example, you can use databases such as ChemSpider to view the UV-Visible, Infrared, and NMR spectra of various chemicals. Although spectroscopic data is related to computerized chemical formats and can be saved in various file formats, it lacks the level of standardization found in the chemical file formats.
SMARTS
While searching databases of chemical structures it is not always the case that the user does have a certain structure to search. For example, medicinal chemists who are synthesizing various derivatives of their molecule would want to know if others have synthesized compounds similar to it. This can be somehow achieved by placing wildcards in the chemical structure such as the R group usually used in chemical sketches. Daylight Chemical Information Systems have developed SMARTS (SMILES Arbitrary Target Specification) for this purpose. SMARTS is a linear notation that is built on SMILES to specify substructures and structural patterns used for querying chemical databases.
Biological Assay Databases
Biological assays are very important in the development of novel drugs. Assays can be used to measure different biological properties of chemicals such as toxicity, anti-inflammatory activity, anticancer activity, or enzyme inhibition activity. These data are usually collected from the literature and stored in databases where other researchers can search for them. The collected data can be used to build predictive models based on an enormous number of experiments that can hardly be done by a single organization alone. The developed models use information from all the previous experiments to build a predictive model that can predict the value of the assay for virtually designed compounds before synthesizing them. We will talk more about these models in the QSAR chapter.
An example of a database that stores biological assays is PubChem. This is an image of the PubChem website:
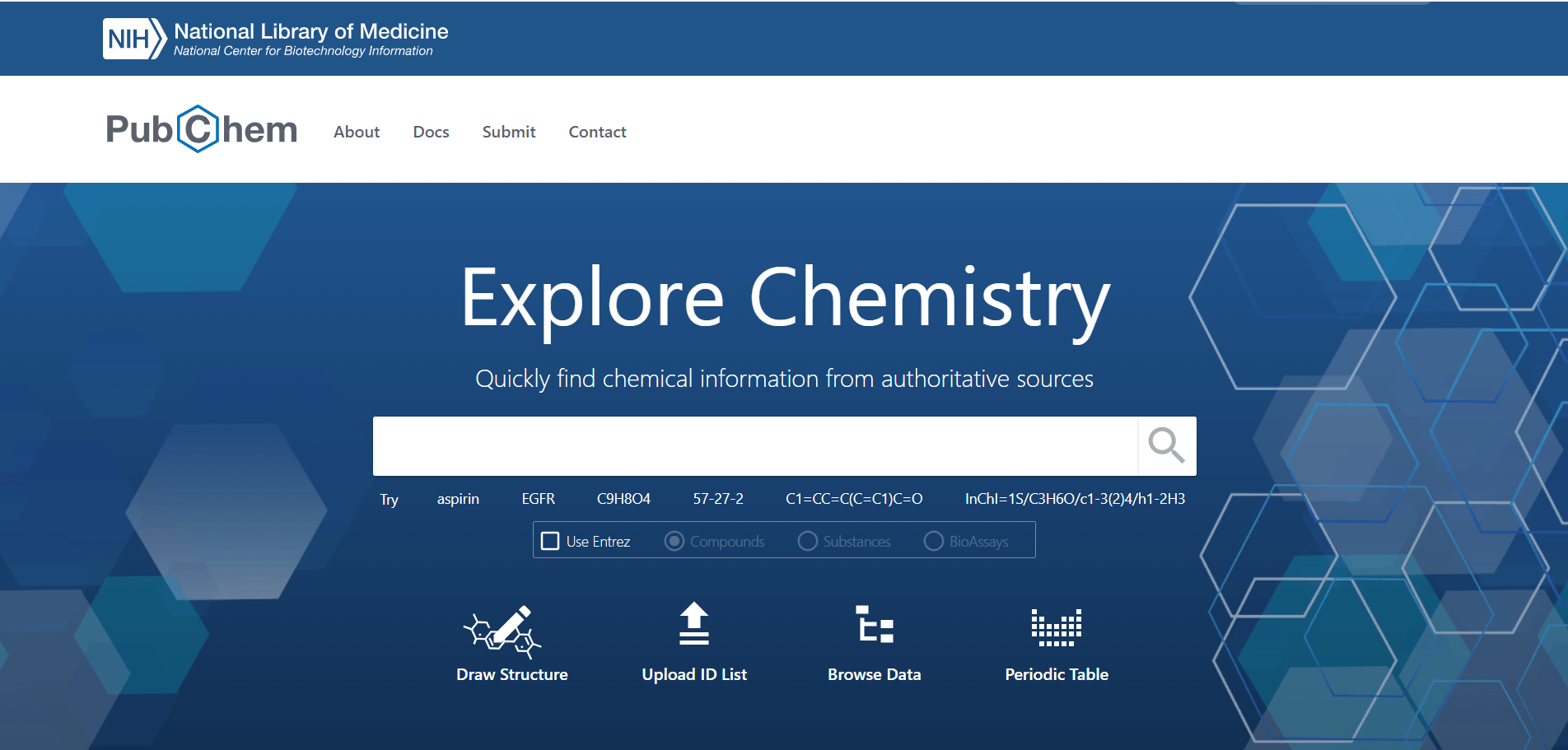
The website, as of the day this chapter was prepared, says that it contains information on 122 million compounds and the values of 297 million bioassays.
You can use the search bar in PubChem to search for a compound using the compound's common name, IUPAC name, CAS number, SMILES, or InChI. Another option is to draw the structure after pressing the Draw Structure button. Here, we searched for haloperidol, a dopamine receptor (D2) antagonist that works as an antipsychotic drug.
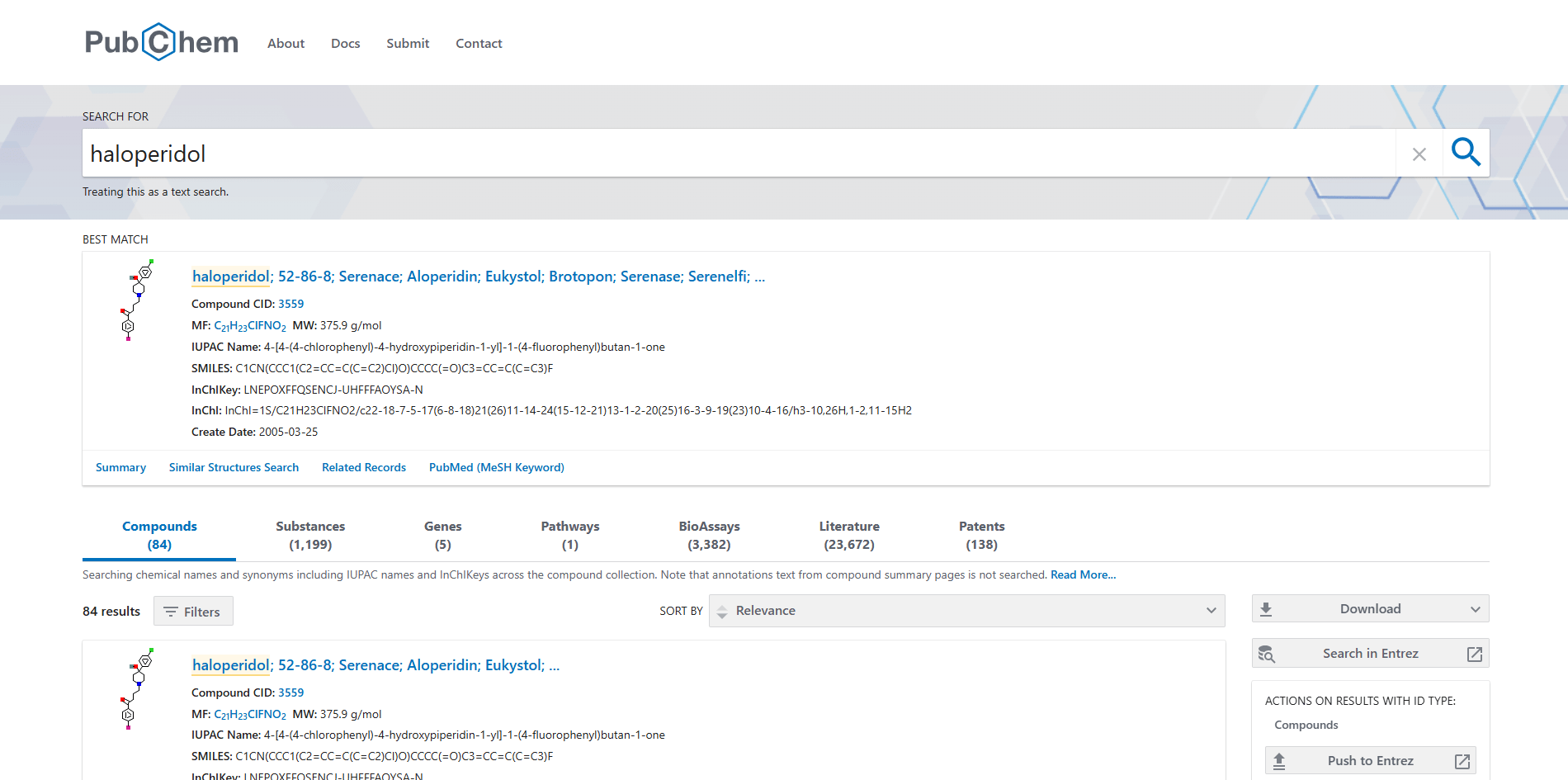
You can see that PubChem showed the best matching compound structure and chemical formats. When clicking on the compound name the following page will be loaded:
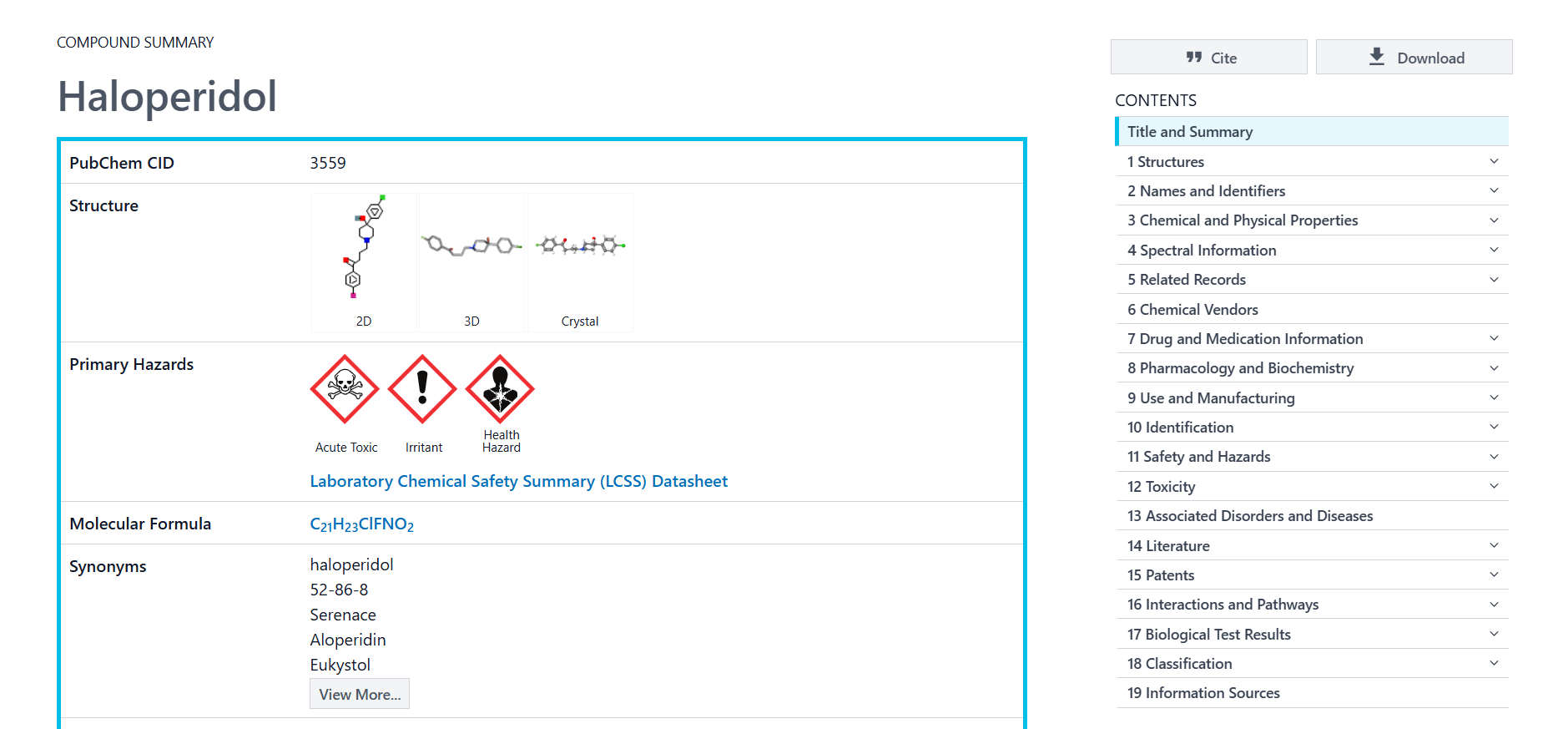
You can see a summary of information about the compound. On the right you will also see a sidebar with many sections about known information of the compound.
Structural Databases
Structural databases are databases dedicated to storing 3D structural information of molecules, ranging from structures of small molecules, as in the Cambridge Structural Database (CSD), to macromolecules such as the Protein Data Bank (PDB). The 3D structure of molecules can generally be elucidated using experimental techniques such as X-Ray Crystallography, NMR, or Cryo-Electron Microscopy. Each of these techniques has advantages and disadvantages. However, X-Ray Crystallography is the technique most useful in chemoinformatics pipelines.
For small molecules, X-Ray Crystallographic structures are typically used as the minimum-energy state of the compound. This is because the technique uses a highly pure and ordered crystal of the compound to determine the relative 3D locations of the compound atoms, where the compound is usually in its minimum-energy state.
For macromolecules, their 3D structure offers unlimited potential for use. The 3D structure can help understand how different amino acid chains interact with each other. Additionally, it can be used in various chemoinformatics techniques such as docking and molecular dynamics. Many researchers are also developing new techniques based on protein structures.
To take a quick glance at how these databases look, we will use the PDB database to search for the protein structure of p53, also known as The guardian of the genome. If you open the PDB website, you will see the following page:
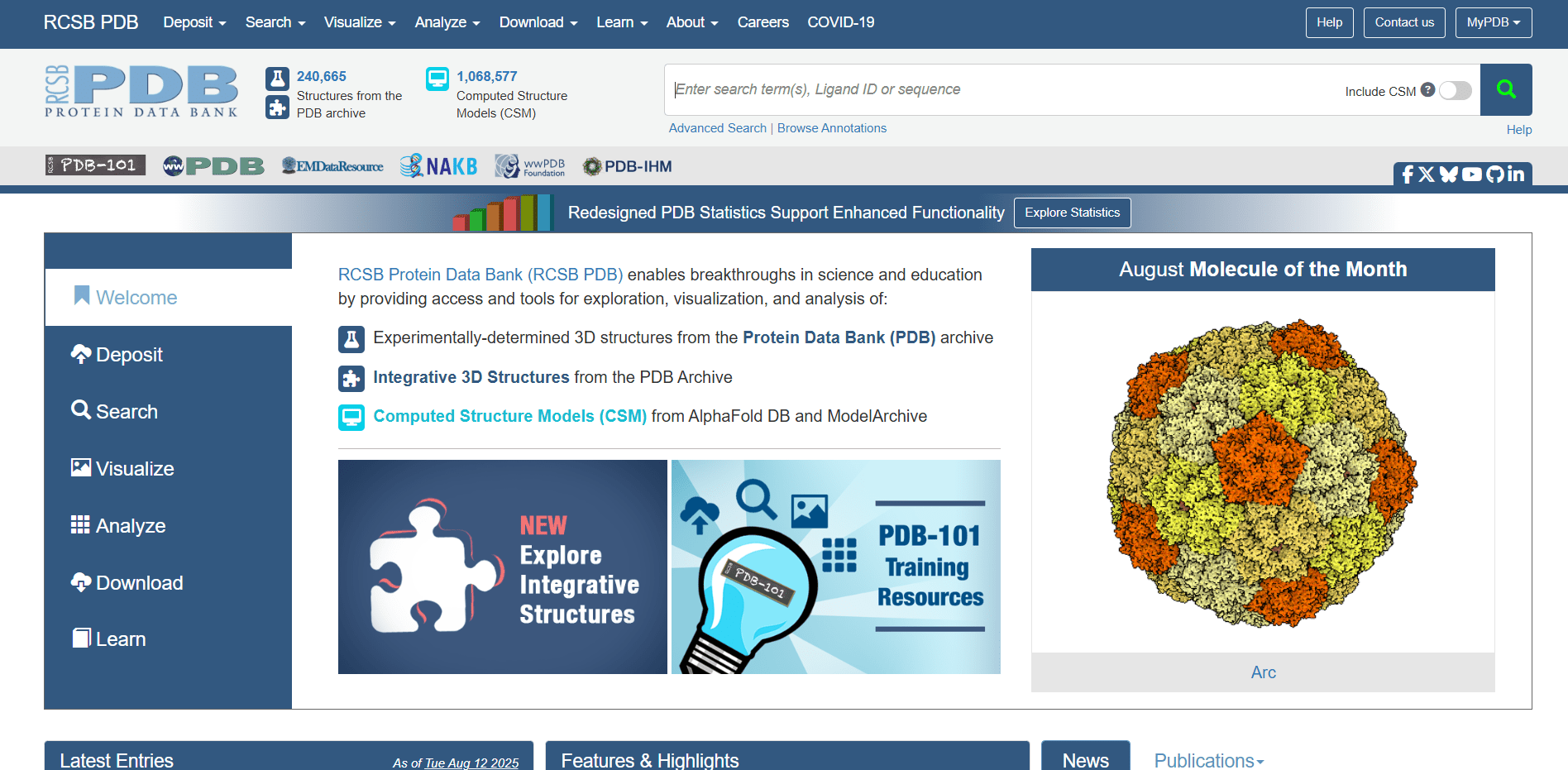
In the header you can see that there is 240 thousand experimentally elucidated structures and is more than one million computed structures in the database. You can also see that there in the database. Write in the search bar p53 and click on search button.
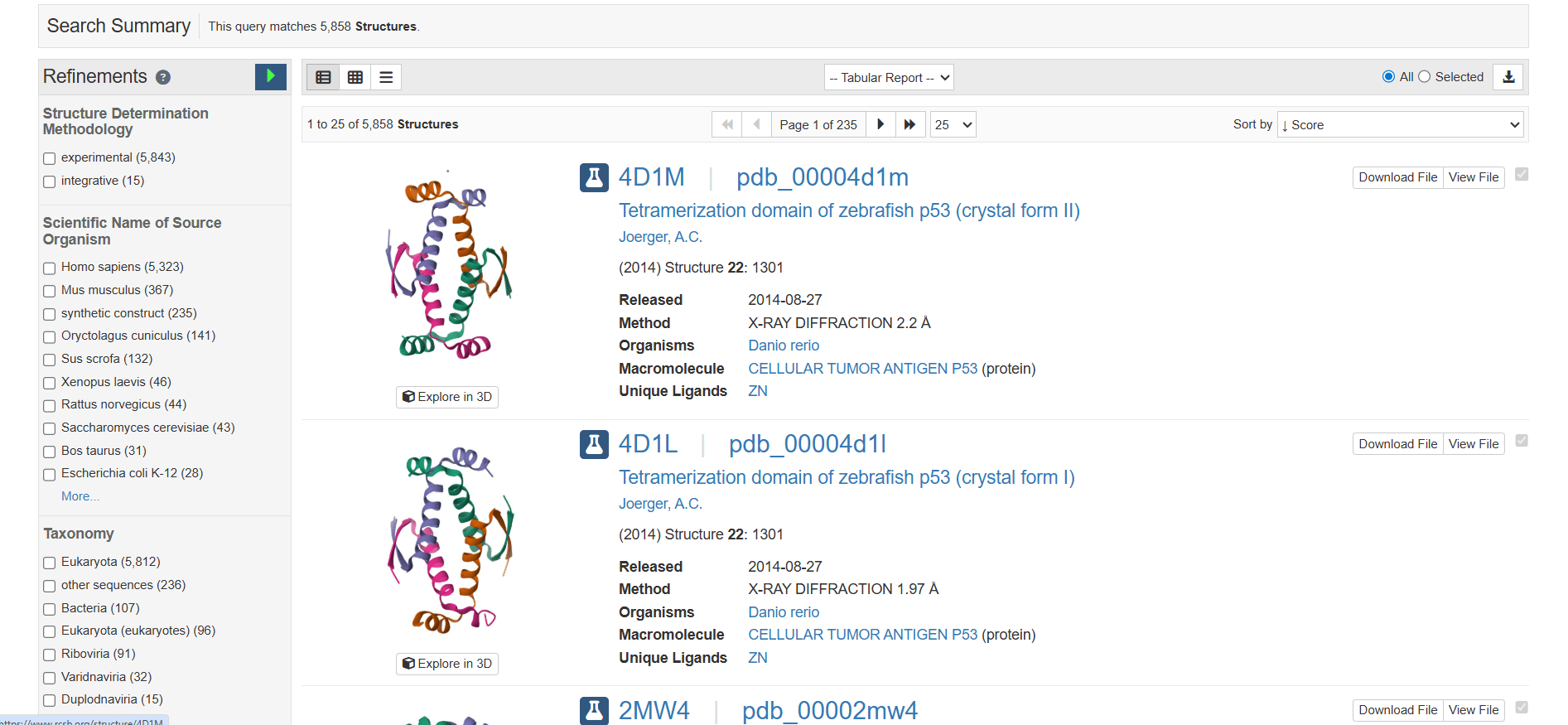
As you can see, there are 5,858 structures with the name p53. You can also see an image of each protein displaying the α-helices and a short title for each structure. The first two proteins are from zebrafish, as the title and the organism's latin name, Danio rerio, indicate. On the left, there are filters you can use to limit the search results to what you actually need. Click on the first protein, and you will see the following page:
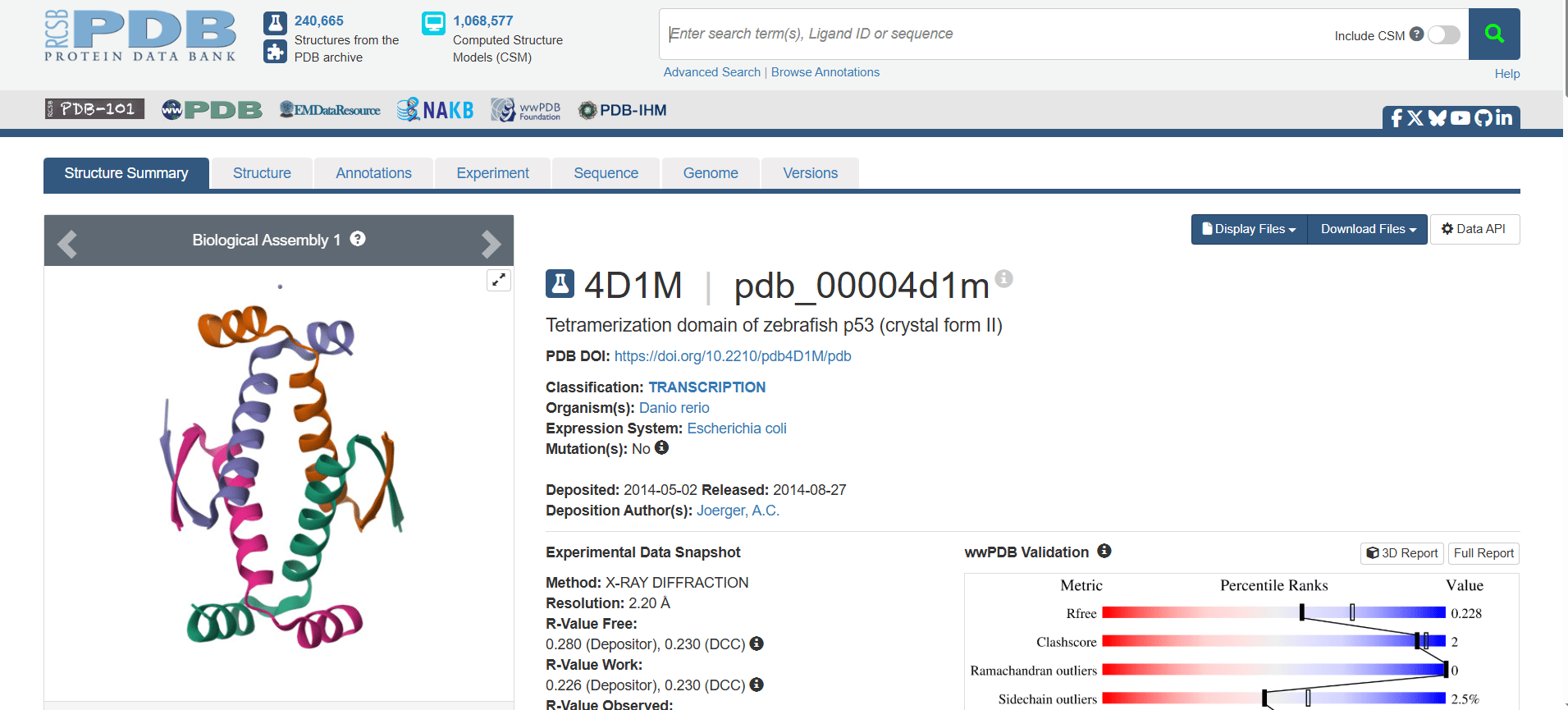
This page is for the protein with the ID 4D1M. Click on the Structure tab at the top to see the 3D structure interactively in the browser, or click on the Download Files button to download the structure in different formats such as PDB.
Due to the difficulties in elucidating 3D structures of proteins, computational tools were developed to predict the complete structure of proteins from their sequence alone. Although these predictions are less accurate than experimental techniques, they are very useful for proteins that do not have a 3D structure deposited in the databases. One of the most popular, accurate, and easy-to-use programs is AlphaFold. This program was developed by Google DeepMind and uses AI models to predict the structure of complex biomolecules containing a mixture of DNA, RNA, protein, ions, and ligands. It has been used to predict over 200 million protein structures, covering nearly all proteins known to science, which are now stored in a freely available database known as the AlphaFold Protein Structure Database. For predicting new structures that are not available in the database, you can use the AlphaFold Server.