In the early stages of drug discovery, medicinal chemists often synthesized numerous derivatives of a chemical scaffold and tested their biological activity. To rationalize which derivatives to prioritize, they identified structural features that were critical for activity. For example, look at the following sketch of fluoroquinolone scaffold:
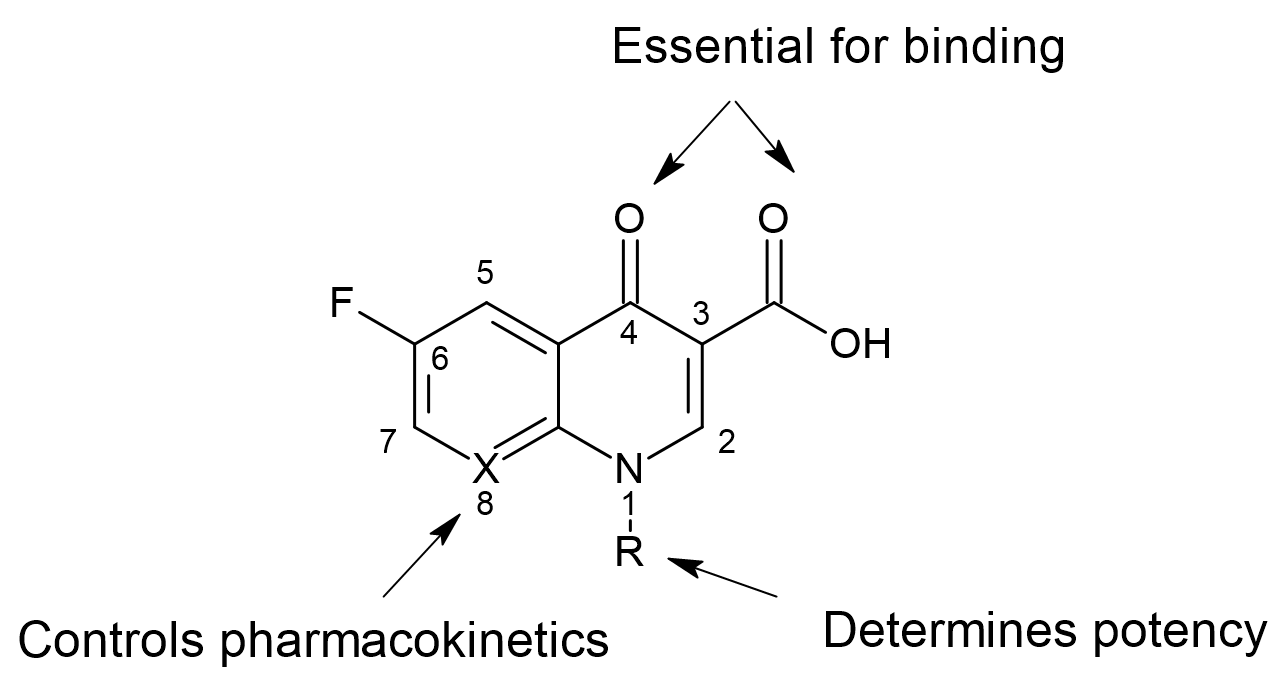
Medicinal chemists traditionally described how structural modifications influence biological activity by referring to the specific positions on the scaffold where substitutions occur. In the fluoroquinolone class of antibiotics for example, functional groups at positions 3, 4, and 6 are essential for antibacterial potency, bulky substituents at position 1 tend to enhance activity, and the atom at position 8 primarily affects pharmacokinetics rather than potency. This is known as Structure Activity Relationship (SAR). However, in order for a molecule to be active on a specific protein, it is not only the chemical structure that matters, but more importantly the interactions it can form with the protein and the physicochemical properties it possesses. This means that molecules with different chemical structures can sometimes produce similar biological effects if they share key interaction patterns and physicochemical characteristics. By understanding and replicating these properties in new derivatives, we can effectively explore chemical space more efficiently and design compounds that maintain or enhance activity.
This concept forms the basis of Quantitative Structure-Activity Relationship (QSAR) modeling. In QSAR, various properties of already synthesized compounds are calculated; these properties are called Descriptors. Descriptors can range from simple 1D properties, such as molecular weight or calculated logP, to more complex 2D and 3D descriptors, which encode information about molecular topology, shape, and electronic distribution. These numerical descriptors provide a way to quantify the moleculer features in a form that can be correlated with biological activity or other target properties.
Once these descriptors are calculated, computational models are developed to link molecular properties to biological activity. These models can then be used to predict the activity of virtually designed compounds before synthesis, significantly reducing experimental workload. For example, a QSAR model could indicate which modifications to a molecule are likely to increase its binding affinity to an enzyme or improve selectivity for a receptor.
For example, look at the following picture of a simple QSAR model that uses one descriptor (LogP) and predicts compound activity (LogIC50) using linear regression.
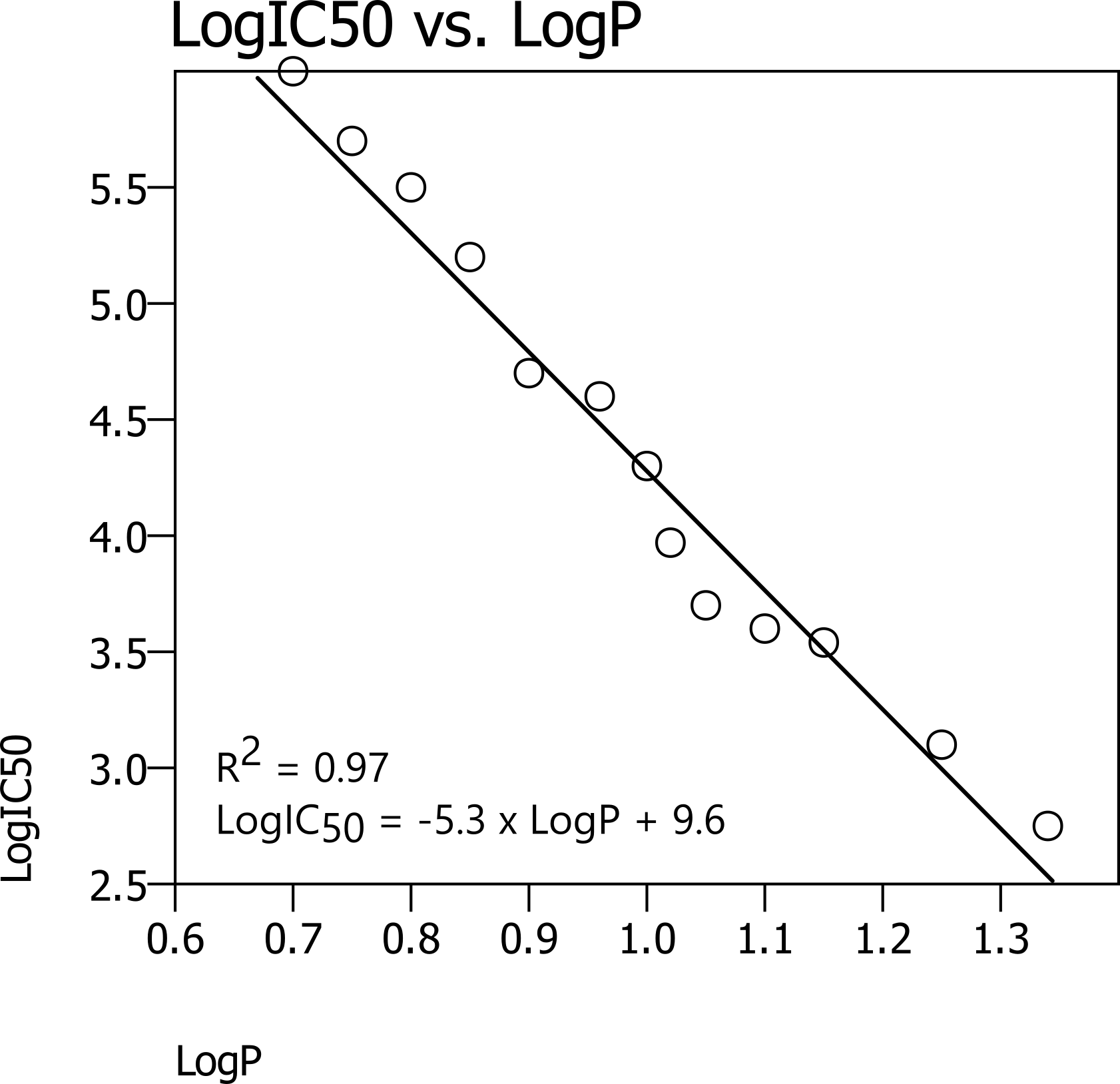
In the image, the x-axis represents LogP, while the y-axis represents LogIC50. The data points correspond to the experimental values used to construct the linear regression QSAR model, and the line represents the plot of the derived QSAR equation. Knowing that LogIC50 is inversely related to potency, it can be observed that higher LogP values are associated with greater potency. When a new derivative is to be evaluated, its LogP descriptor can be calculated and substituted into the equation to predict the corresponding LogIC50. This is a simple example of how QSAR works. However, in real case scenarios, hundreds or even thousands of descriptors are used in building QSAR models.
Types of QSAR Descriptors
Descriptors are the numerical representation of molecular properties that can be used to predict biological activity. They are typically classified as:
-
1D Descriptors
Simple molecular properties calculated from the chemical formula or basic structure.
Examples: molecular weight, number of hydrogen bond donors/acceptors, calculated logP (lipophilicity), number of oxygen atoms.
These descriptors are useful for modeling general trends in activity and are easy to interpret.
-
2D Descriptors
Encodes the moleculer topology and connectivity without requiring 3D information.
Examples: Topological indices, Kappa Shape Indices, and Wiener index
-
3D Descriptors
Describes the three-dimensional shape of the molecule.
QSAR Modeling algorithms
QSAR models can be constructed using a variety of statistical and machine learning algorithms, each offering a different balance between interpretability and predictive power. Traditional approaches such as Multiple Linear Regression (MLR) or Partial Least Squares (PLS) are valued for their transparency, allowing researchers to clearly understand how each descriptor influences the predicted activity. However, as chemical datasets grow in complexity, more sophisticated non-linear methods like Support Vector Machines (SVM), Random Forests (RF), and k-Nearest Neighbors (k-NN) have gained prominence due to their ability to capture intricate relationships between molecular descriptors and biological activity. We will talk more about QSAR modeling in a dedicated course.